Ravy Ieng (2022)
![]()
![]()
|
|
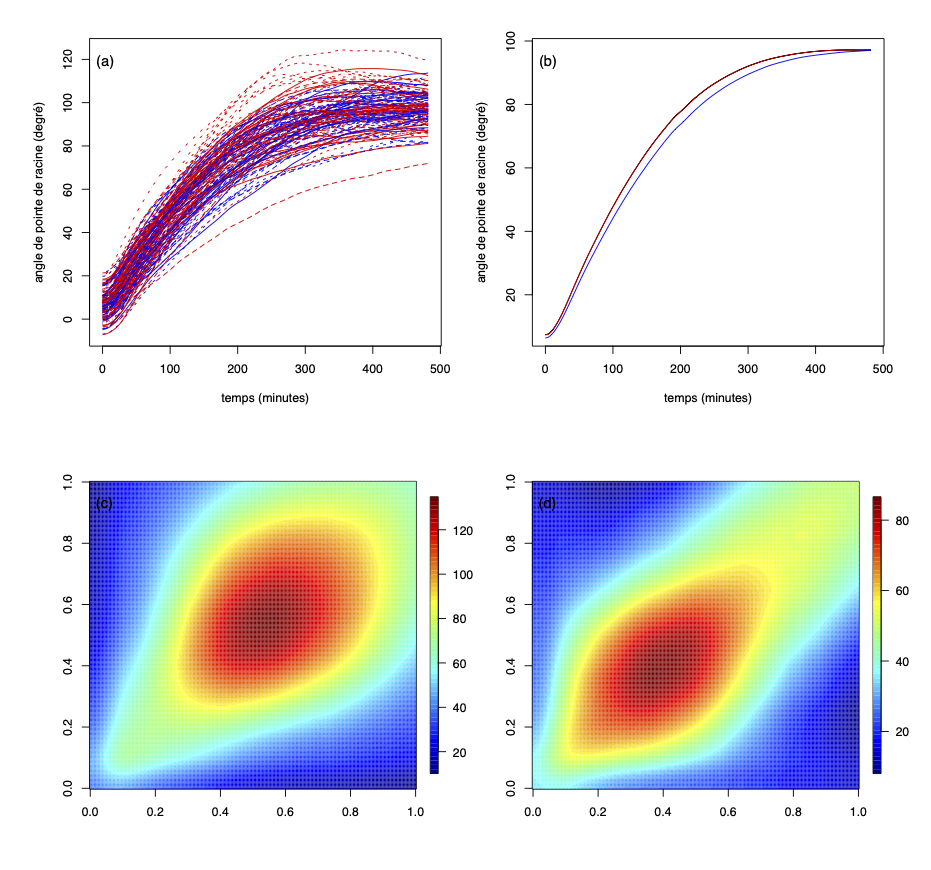
Association génétique mesurée à l'aide d'une méthode de classification de données fonctionnelles. Ce mémoire propose une nouvelle méthode d’association génétique quand le caractère génétique d’intérêt est une fonction. À cette fin, nous présentons une méthode non-paramétrique de classification supervisée pour des données fonctionnelles, développée par Ferraty et Vieu (2006). Dans un premier temps, nous présentons quelques concepts de l’analyse de données fonctionnelles et passons en revue le problème de classification dans le cas fonctionnel, la méthode des K plus proches voisins et les fonctions noyaux. Nous introduisons trois familles de semi-métriques qui agissent comme une mesure de proximité entre deux fonctions. Par la suite, nous étudions la performance de la méthode non-paramétrique de classification supervisée à l’aide d’une étude de simulation. Finalement, nous présentons l’approche consistant à utiliser la méthode de classification non-paramétrique aux données génétiques d’une plante à fleurs (appelée arabette des dames) de Moore et al. (2013) afin d’étudier l’association entre le phénotype et la lignée d’origine de la plante. On compare les résultats observés avec les résultats d’une approche par vraisemblance de Kwak et al. (2016). On constate que la méthode proposée donne des résultats différents de l’approche par vraisemblance |
|
|
|
Renaud Alie (2020)
![]()
![]()
|
|
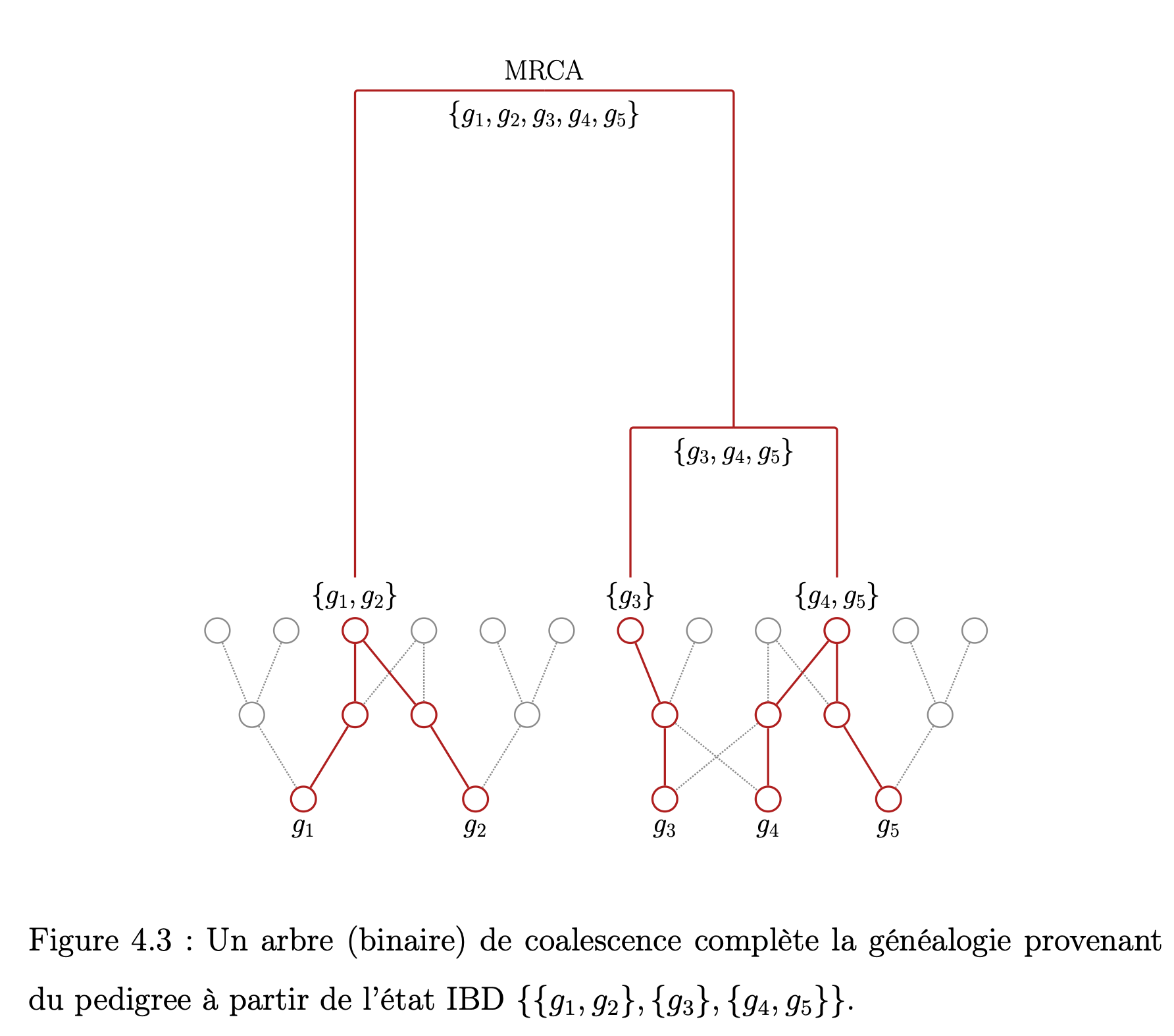
Le p-coalescent : un modèle probabiliste intégrant la génétique familiale au processus de coalescence. L'objectif central de ce mémoire est de présenter une approche qui permet d'inclure les contraintes relevant de la génétique familiale et des pedigrees dans un modèle de génétique des populations inspiré du processus de coalescence. Ce modèle est formulé en termes de graphes dirigés acycliques et, plus précisément, d'arbres appelés généalogies. Quelques notions fondamentales concernant le modèle de Wright-Fisher et la génétique familiale sont d'abord formulées. Le modèle proposé, le p-coalescent, est ensuite présenté avec quelques résultats concernant le comportement des distances sur le graphe et la relation qui existe entre la taille de la généalogie et le nombre de mutations observées. |
|
|
|
Na Yang (2019)
![]()
![]()
|
|
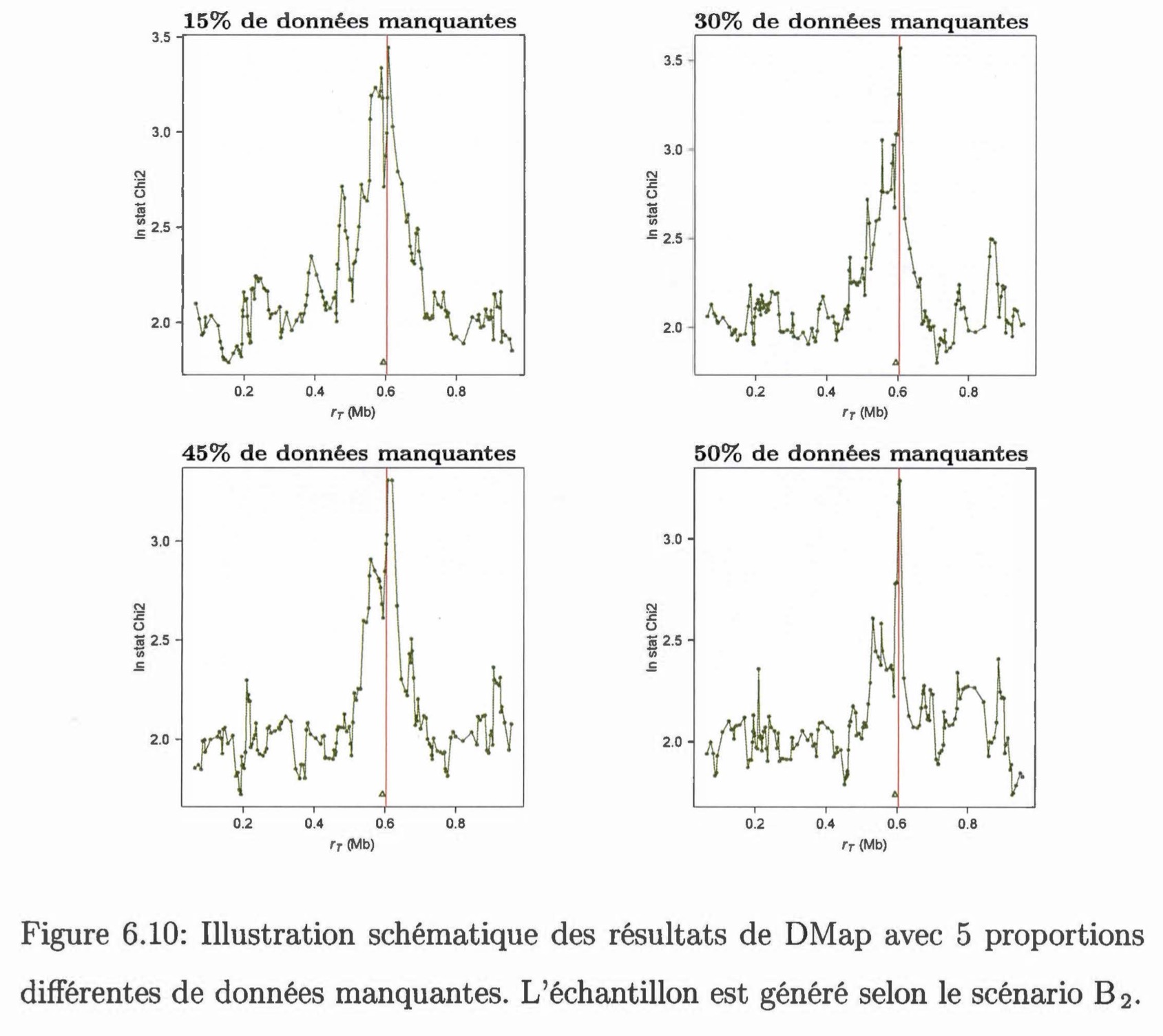
Comparaison entre une méthode de cartographie fine et des tests d'association dans le cadre de l'étude d'un caractère génétique avec des données incomplètes. Nous présentons et testons deux méthodes permettant d'estimer la position d'une mutation causale le long d'une séquence génétique dans un échantillon en présence de données manquantes. La première méthode, DMap, s'insère dans la méthodologie de la cartographie génétique fine, basée sur le processus de coalescence. Cette méthode est capable d'analyser des données incomplètes directement en les considérant non-informatives. La deuxième méthode est un test d'association qui utilise des données imputées par la méthode IMPUTE. Ce type de test (SNPTEST) est beaucoup appliqué dans les études d'association sur le génome. On compare ces deux méthodes par une étude de simulation selon plusieurs scénarios. On constate que la méthode DMap a une performance comparable à la méthode SNPTEST. Si environ 50% de données sont manquantes dans l'échantillon, il sera plus avantageux d'utiliser la méthode DMap.
|
|
|
|
Éric Marcotte (2018)
![]()
![]()
|
|
Une nouvelle approche computationnelle pour la génération de graphes de recombinaison ancestraux comportant un grand nombre de marqueurs génétiques. Nous présentons dans ce mémoire une nouvelle approche permettant de créer et d'exploiter des graphes de recombinaison ancestraux comportant un grand nombre de marqueurs génétiques. Nous utilisons principalement une représentation binaire ainsi que des fonctions booléennes applicables sur des séquences de marqueurs afin d'implémenter un logiciel fait sur mesure pour accomplir cette tâche de manière efficiente. De plus, nous détaillons une heuristique de construction bien connue et nous proposerons un algorithme novateur minimisant le nombre de recombinaisons requises pour parvenir à un ancêtre commun. Enfin, nous exposons quelques résultats sur l'efficacité de notre approche.
|
|
|
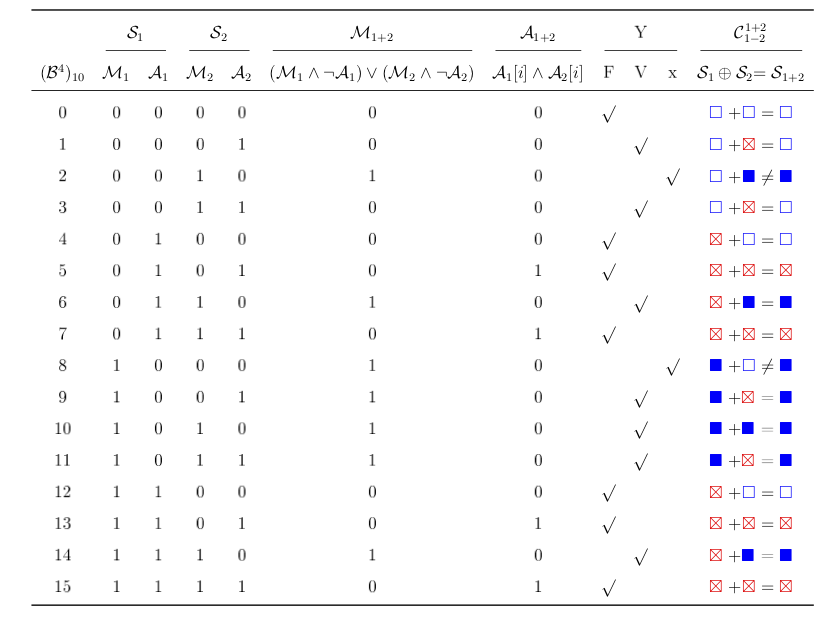
La table de vérité pour le calcul d'une coalescence stricte. Le calcul de l'ensemble A1+2 correspond à un et logique sur les séquences A d'origine. On peut voir aussi la correspondance symbolique utilisée pour représenter l'encodage binaire.
|
Myriam Ziou (2017)
![]()
![]()
|
|
Cartographie de caractères polygéniques par le processus de coalescence. Ce mémoire introduit une adaptation à une méthode de cartographie génétique existante à des traits causés par deux mutations. Nous commençons par énoncer certains concepts importants en génétique, puis nous présentons des méthodes de cartographie génétique existantes. Nous introduisons par la suite le processus de coalescence et un de ses dérivés important : le graphe de recombinaison ancestral. Cela nous permet de présenter la méthode sur laquelle on s’est basé ainsi que la nôtre, qui utilisent toutes deux ce type de graphe. Nous finissons par simuler des données génétiques afin de tester notre nouvelle méthode sur celles-ci en faisant varier plusieurs paramètres, ce qui nous permet de déterminer ses forces comme ses limitations.
|
|
|

|
Sadoune Ait Kaci Azzou (2016)
![]()
![]()
|
|
Estimation de l'historique démographique d'une population de virus à partir de séquences d'adn par la théorie de coalescence.L'évolution de la taille d'une population peut être retracée à partir d'un échantillon de séquence d'ADN. Dans cette thèse, nous proposons une nouvelle méthodologie non paramétrique basée sur une stratégie d'échantillonnage pondéré (Importance Sampling) qui permet d'explorer de tels historiques démographiques. L'essence de la méthode est de simuler un grand nombre de généalogies en utilisant le processus de coalescence, où l'information fournie par ces généalogies est combinée en utilisant les poids de cet échantillonnage pondéré. En premier, nous proposons la méthode skywis plot qui débute par l'estimation de la taille de la population effective pour chaque généalogie, pour chaque intervalle de temps prédéfini, appelé époque; ensuite, une moyenne pondérée de ces tailles de population estimées est calculée. Ainsi, les généalogies qui sont le plus en accord avec les données ont un poids plus élevé. Nous avons aussi généralisé notre méthodologie au cas d'un échantillonnage en série. Cela a nécessité la mise en œuvre d'une stratégie d'échantillonnage efficace qui permet de tenir compte de cette réalité qui est très utilisée, notamment dans le cas de virus qui évoluent rapidement comme le VIH. Ensuite, nous proposons d'améliorer la performance de la méthode skywis plot à travers une procédure itérative appelée iterative calibrated skywis plot; la taille de la population effective est approximée par une fonction en escalier, qu'on ré-estime après chaque itération en utilisant la méthode calibrated skywis plot. Ces fonctions en escalier sont utilisées pour générer les temps d'attente d'un processus de Poisson non homogène (coalescence avec mutation) sous un modèle avec une taille de population variable. Cela nous a aussi amené à adapter la distribution proposée de Stephens et Donnelly (2000).
|
|
|
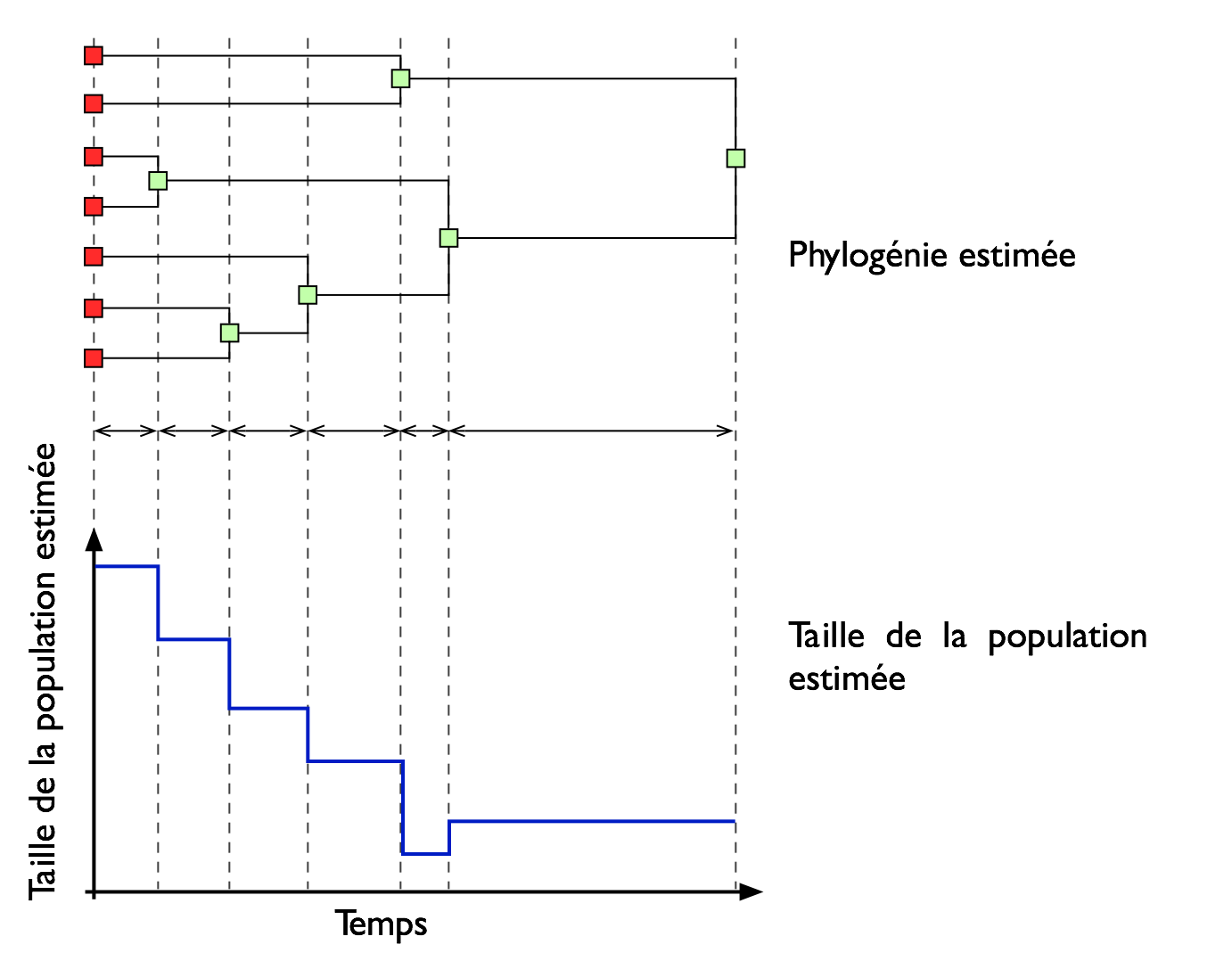
La recherche consiste à estimer l'histoire de la taille de la population à partir de la généalogie de celle ci, généalogie qui est construite à partir des séquences d'ADN. |
Abdelhakim Ferradji (2016)
![]()
![]()
|
|
GOLIATE : un nouveau test d’association génétique combinant entre le processus de coalescence et les modèles linéaires mixtes. Dans ce mémoire nous présentons un nouveau test d'association génétique qui permet d'analyser simultanément un ensemble de SNPs d'une région chromosomique, tout en tenant compte d'une éventuelle présence de structure de population dans l'échantillon d'étude. Ce test est basé sur un modèle linéaire mixte qui capture l'effet de la structure de population grâce à une nouvelle matrice de similarité, construite en utilisant le processus de coalescence avec recombinaison. Des simulations sont effectuées pour tester et comparer les performances de notre nouvelle approche avec quelques tests proposés dans la littérature. Notre test montre un bon contrôle de l'erreur de type 1 en présence de structure de population contrairement aux autres tests, et semble avoir une puissance comparable à celle des autres méthodes dans le cas des variants génétiques rares.
|
|
|
|
Cédric Beaulac (2015)
![]()
![]()
|
|
Intelligence artificielle avec apprentissage automatique pour l'estimation de la position d'un agent mobile en utilisant les modèles de markov cachés. Dans ce mémoire, nous développons une méthodologie afin d'estimer la position d'un agent mobile dans un environnement borné. Nous utilisons un modèle de semi-Markov caché pour modéliser notre problématique. Dans ce contexte, l'état caché est la position de l'agent. Nous développons les algorithmes et programmons une intelligence artificielle capable de faire le travail de façon autonome. De plus, nous utilisons l'algorithme de Baum-Welch pour permettre à cette intelligence d'apprendre de ses expériences et de fournir des estimations plus précises au fil du temps. Finalement, nous construisons un jeu vidéo pour mettre à l'épreuve cette intelligence artificielle. Le fonctionnement de la méthode est illustré avec plusieurs scénarios, et nous montrons que la méthode que nous proposons est meilleure que d'autres.
|
|
|
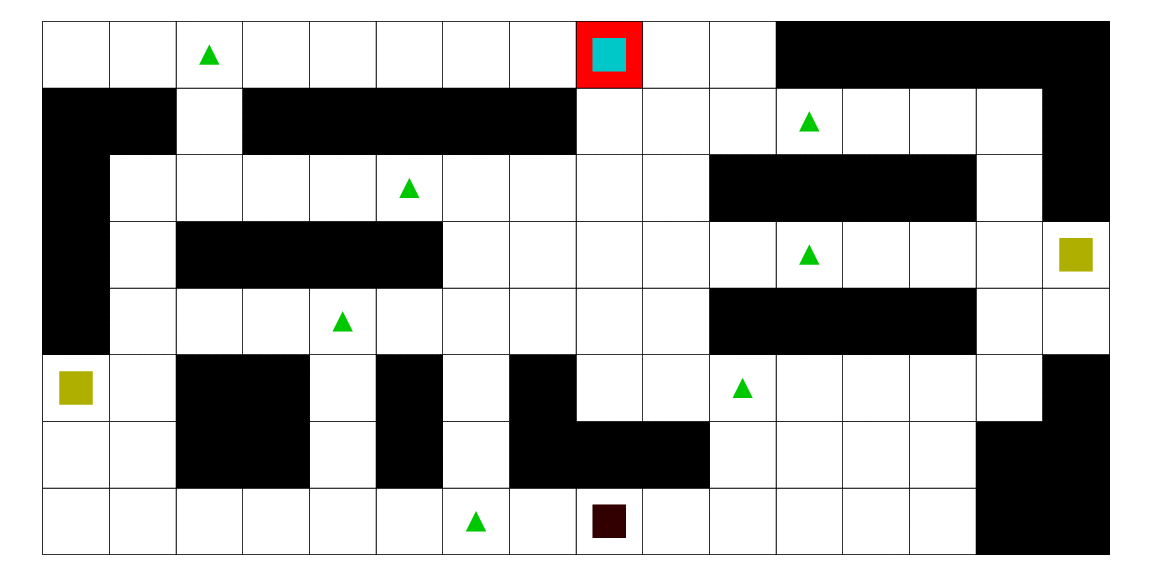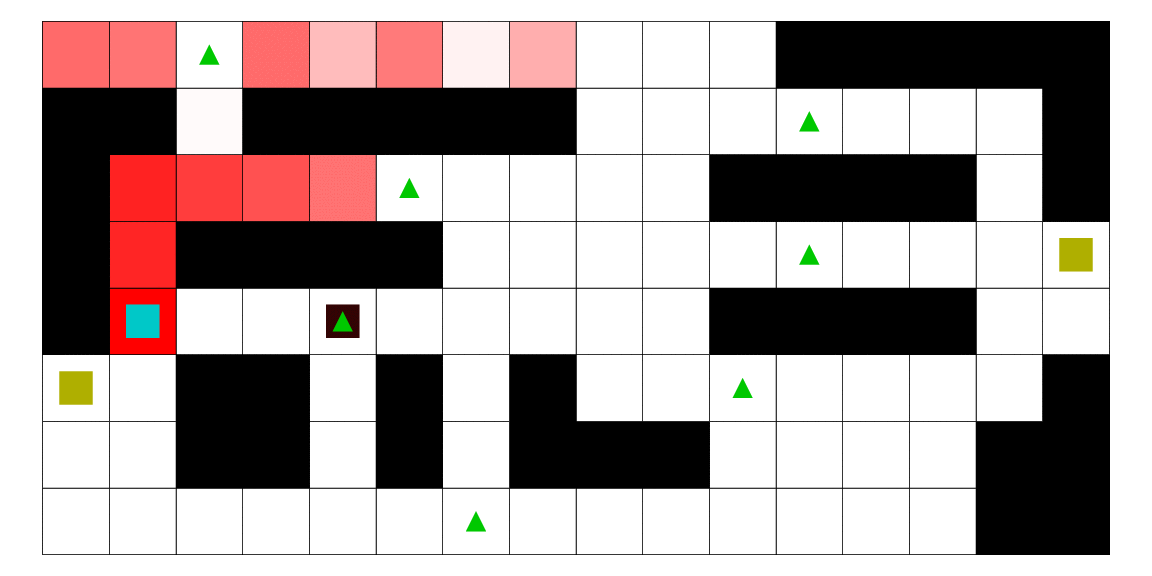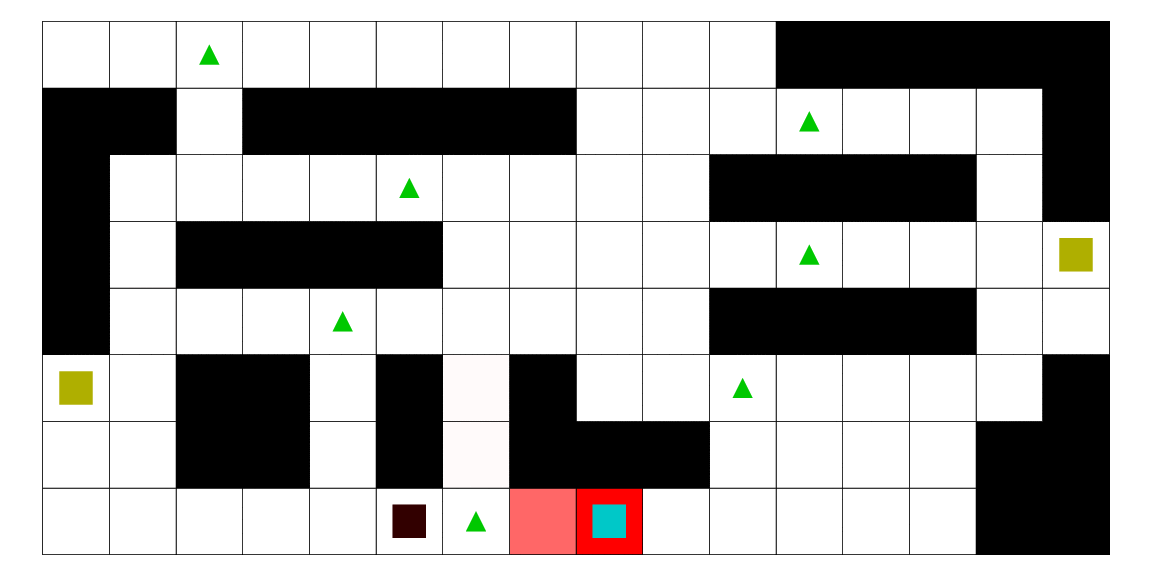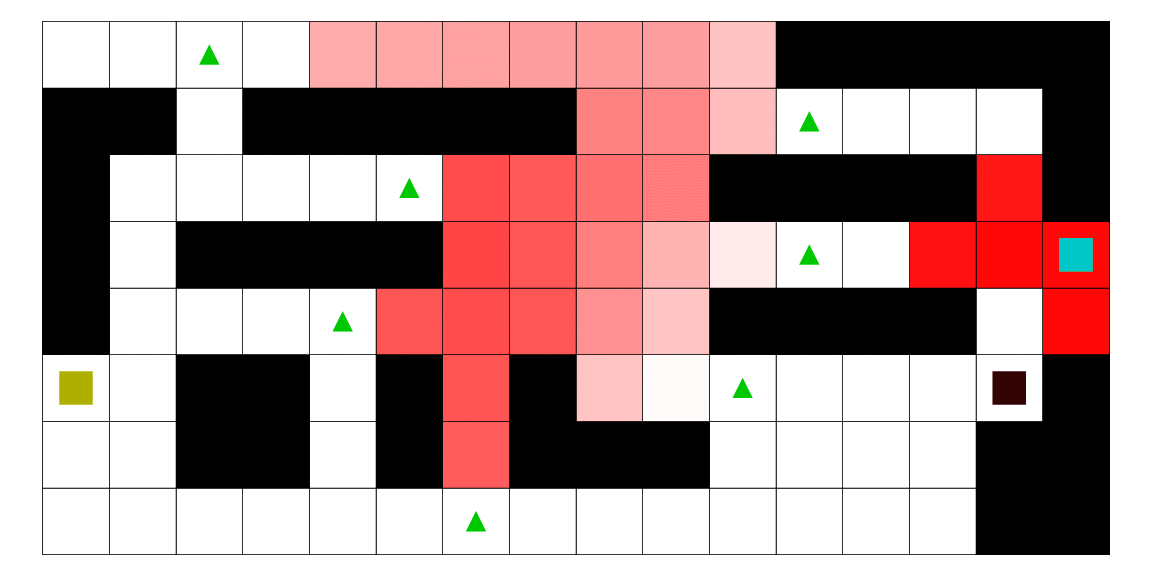
Le pirate (carré noir), qui a une vue limitée à une case autour de lui, cherche à attraper le matelot (carré bleu). Le matelot, lui, cherche à aller vers un des deux trésors (carrés jaunes). À chaque déplacement, le pirate se déplace en fonction des probabilités estimées de la position du matelot; les teintes rouges des cases sont proportionnelles à la probabilité estimée que le matelot se trouve dans cette case. Ces probabilités sont estimées à l'aide d'un modèle de semi-Markov caché, avec apprentissage
|
Oscar Camillo Ortiz (2015)
![]()
|
|
Analyse par grappe de mesures de cytométrier pa flux à des fins de caractérisation de patients cancéreux. La Cytométrie par Flux (CF) est un outil très important pour identifier des composants de cellules. Cette analyse est effectuée par l'utilisation des fluorescents (variables). Cependant, l'analyse CF nous permet d'analyser, au maximum, la distribution des cellules uniquement par paires de variables en utilisant des graphiques de dispersion (scatter plots). Compte tenu de cette limite, il est difficile de tenir compte de la corrélation entre toutes les variables. Cette difficulté peut être surmontée à travers l'analyse par grappes qui tient compte des relations ( corrélations entre les variables). Dans ce rapport nous avons utilisé la méthodogie de l'analyse par grappes pour arriver à des conclusions concernant les cellules, en tenant compte de toutes les variables en même temps. Après avoir identifié les groupes de cellules, nous allons nous baser sur ces groupes de cellules pour identifier les groupes de patients qui ont des caractéristiques similaires. Cette méthodologie peut donc s'avérer utile pour aider à identifier plus facilement les patients qui ont des caractéristiques similaires, en tenant compte de l'ensemble des cellules en même temps.
|
|
|

|
Mathieu Dupont (2013)
![]()
![]()
|
|
Cartographie génétique fine : évaluation d’une méthode d’estimation des allèles et du modèle de pénétrance. Nous présentons et testons une méthode d’estimation des allèles d’une mutation potentiellement associée à un phénotype ainsi qu’une méthode d’estimation de son modèle de pénétrance. Ces deux méthodes reposent sur un algorithme EM et s’insèrent dans une mé- thode de cartographie génétique fine, MapARG, basée sur le processus de coalescence. La sensibilité des deux méthodes d’estimation aux risques relatifs du modèle de pénétrance réel de la mutation, à la taille des échantillons ainsi qu’à la largeur des fenêtres utilisées est systématiquement évaluée. Les deux méthodes s’avèrent performantes, particulièrement pour des risques relatifs forts. La taille des échantillons exerce peu d’influence, mais des fenêtres plus larges donnent de meilleurs résultats. L’estimation préalable du modèle de pénétrance montre un certain effet bénéfique sur l’estimation subséquente des allèles, comparativement à l’utilisation du vrai modèle connu. Aussi, la méthode d’estimation du modèle de pénétrance, basée sur une distance calculée entre les haplotypes primitifs et mutants, montre en soi un certain potentiel comme méthode de cartographie génétique.
|
|
|
|
Marie-Hèlène Descary (2012)
![]()
![]()
|
|
DMAP : une nouvelle méthode de cartographie génétique fine adapteé à des modèles génétiques complexes. Dans ce mémoire, nous présentons une nouvelle méthode de cartographie génétique fine ayant comme particularité de pouvoir être utilisée dans le cadre de modèles génétiques complexes. Nous présentons tout d’abord quelques concepts de génétique et de statistique génétique, avec une emphase particulière sur le processus de coalescence qui est à la base de notre travail. Par la suite, trois méthodes de cartographie génétique déjà existantes sont présentées; notre nouvelle méthode contient des éléments de chacune d’entre elles. Nous décrivons ensuite la nouvelle méthode proposée dans ce mémoire. Finalement, nous testons notre nouvelle approche à l’aide de simulations; nous comparons par la suite les résultats obtenus avec notre méthode à ceux obtenus par des tests d’association classiques, et par deux des trois méthodes présentées au début du mémoire. Les résultats nous laissent croire que notre méthode est performante autant dans des cas de modèles génétiques simples que complexes, contrairement à la plupart des méthodes existantes.
|
|
|

|
Marie Forest (2010)
![]()
![]()
|
|
Cartographie génétique fine simultanée de deux gènes. Dans le domaine de la recherche de gènes causaux, il est maintenant connu que plusieurs caractères complexes peuvent en fait être influencés par une multitude de gènes. Dans ce mémoire, nous présentons l’adaptation d’une méthode de cartographie génétique fine à la cartographie de caractère polygénique. Nous présentons tout d’abord un aperçu de certains outils statistiques utilisés en génétique. En particulier, certaines mesures d’association généralement employées en cartographie génétique. Puis, nous présentons la méthode de cartographie que nous souhaitons adapter : méthode qui suppose que le caractère est causé par l’effet d’un seul gène. Nous supposons plutôt que le caractère est causé par la combinaison de deux gènes. Après avoir présenté notre modélisation et les aspects théoriques de l’adaptation proposée, nous utilisons des données simulées pour tester nos développements. Nous comparons aussi nos résultats avec ceux obtenus avec une mesure d’association, ainsi qu’avec la méthode de cartographie dont nous proposons une adaptation. Les résultats démontrent la nécessité de développer des méthodes de cartographie génétique adaptées aux caractères polygéniques ; avec quelques améliorations concernant l’inférence des génotypes aux gènes causaux, notre adaptation devrait offrir de meilleurs résultats que les autres méthodes présentées.
|
|
|
|
Gabrielle Boucher (2009)
![]()
![]()
|
|
Intégration de la réalité diploïde et des modèles de pénétrance à une méthode de cartographie génétique fine. Nous présentons dans ce mémoire des outils permettant de généraliser une méthode de cartographie génétique fine. Nous y résumons les concepts de base de la statistique génétique et y décrivons aussi la méthode de cartographie génétique fine que nous cherchons à généraliser en permettant l’utilisation de génotypes plutôt que d’haplotypes. Pour ce faire, nous comparons diverses méthodes reconnues d’estimation d’haplotypes. Le développement nouveau de ce travail consiste en un algorithme EM conditionnel aux phénotypes permettant d’estimer les haplotypes associés à un échantillon de génotype, ainsi que le statut au gène causal du caractère étudié. Nous généralisons la méthode de cartographie par l’ajout d’étapes au modèle d’échantillonnage pondéré. Nous effectuons finalement quelques tests par simulation.
Mots-clés : algorithme EM, cartographie génétique, coalescence, diplotype, échantillonnage pondéré, estimation, génotype, gène causal, haplotype, modèle de pénétrance, phénotype, vraisemblance composite.
|
|
|

|
Hugues Massé (2008)
![]()
![]()
|
|
Exploration d'une nouvelle méthode d'estimation dans le processus de coalescence avec recombinaison. L’estimation de paramètres génétiques est un problème important dans le domaine de la génétique mathématique et statistique. Il existe plusieurs méthodes s’attaquant à ce problème. Certaines d’entre elles utilisent la méthode du maximum de vraisemblance. Celle-ci peut être calculée à l’aide des équations exactes de Griffiths-Tavaré, équations de récurrence provenant du processus de coalescence.
Il s’agit alors de considérer plusieurs histoires possibles qui relient les données de l’échantillon initial de séquences d’ADN à un ancêtre commun. Habituellement, certaines des histoires possibles sont simulées, en conjonction avec l’application des méthodes Monte-Carlo. Larribe et al. (2002) utilisent cette méthode (voir chapitre IV). Nous explorons une nouvelle approche permettant d’utiliser les équations de Griffiths-Tavaré de fa ̧con différente pour obtenir une estimation quasi exacte de la vraisemblance sans avoir recours aux simulations. Pour que le temps de calcul nécessaire à l’application de la méthode demeure raisonnable, nous devons faire deux compromis majeurs. La première concession consiste à limiter le nombre de recombinaisons permises dans les histoires. La seconde concession consiste à séparer les données en plusieurs parties appelées fenêtres. Nous obtenons ainsi plusieurs vraisemblances marginales que nous mettons ensuite en commun en appliquant le principe de vraisemblance composite. à l’aide d’un programme écrit en C++, nous appliquons notre méthode dans le cadre d’un problème de cartographie génétique fine ou` nous voulons estimer la position d’une mutation causant une maladie génétique simple.
Notre méthode donne des résultats intéressants. Pour de très petits ensembles de données, nous montrons qu’il est possible de permettre un assez grand nombre de recombinaisons pour qu’il y ait convergence dans la courbe de vraisemblance obtenue. Aussi, il est également possible d’obtenir des courbes dont la forme et l’estimation du maximum de vraisemblance sont similaires à celles obtenues avec la méthode de Larribe et al. Cependant, notre méthode n’est pas encore applicable dans son état actuel parce qu’elle est encore trop exigeante en termes de temps de calcul.
Mots-clés : équations exactes de Griffiths-Tavaré, paramètres génétiques, processus de coalescence, vraisemblance composite.
|
|
|

|
Sarah Vahey (2008)
![]()
![]()
|
|
Intégration de la réalité diploïde et des modèles de pénétrance à une méthode de cartographie génétique fine. Les méthodes de cartographie fine sont des modèles qui estiment la position d’un allèle mutant peuvant causer une maladie dans un groupe d’individus. Le travail de Larribe et al. (2002, 2003), MapArg, n’a pas tenu compte des paramètres de pénétrance jusqu’à maintenant. Ce mémoire démontre les effets de ces paramètres, soient la pénétrance et la phénocopie, sur la performance de MapArg, dans des populations haplo ̈ıdes. De plus, deux méthodes que nous avons développées seront ensuite incorporées à MapArg dans le but d’améliorer son efficacité si il y a pénétrance et/ou phénocopie.
Les résultats démontrent que la phénocopie peut avoir une influence négative sur l’efficacité de MapArg. La pénétrance ne semble pas avoir d’effet majeur sur MapArg. La première méthode développée est un modèle simple qui n’apporte pas d’amélioration majeur de MapArg par rapport à ce mˆeme modèle sans ajustement. Par contre, cela procure un point de départ pour les développements futurs dans les populations diploïdes. La deuxieme méthode améliore l’efficacité de MapArg sous certaines conditions, en particulier, si la taille de l’échantillon est assez grande. La deuxieme méthode fonctionne également très bien pour les données réelles de la Fibrose Kystique (Kerem et al., 1989). Mots clés: phénocopie, pénétrance, pénétrance incomplète, cartographie fine.
Fine mapping methods are models that provide an estimate for locating a mutation causing a given disease among a group of individuals. MapArg, the work of Larribe et al. (2002, 2003), did not take penetrance parameters into account to date. This thesis shows the effect of these parameters, namely penetrance and phenocopy, on the performance of MapArg for haploid populations. Also, two different methods are developed and incorporated into the MapArg framework with the goal of increasing efficacy of MapArg in the presence of penetrance and/or phenocopy. Results show that phenocopy can strongly effect MapArg’s efficiency while penetrance does not have much of an effect. The first Method developed is a simple model that does not prove much more efficient than MapArg without any adjustment; however, it provides the groundwork for further development when diploid populations will be modeled. Method 2 has shown to improve the efficiency of MapArg under certain conditions, in particular, when the sample size is large. This method also greatly improves the performance of MapArg with the Cystic fibrosis data (Kerem et al., 1989).
|
|
|
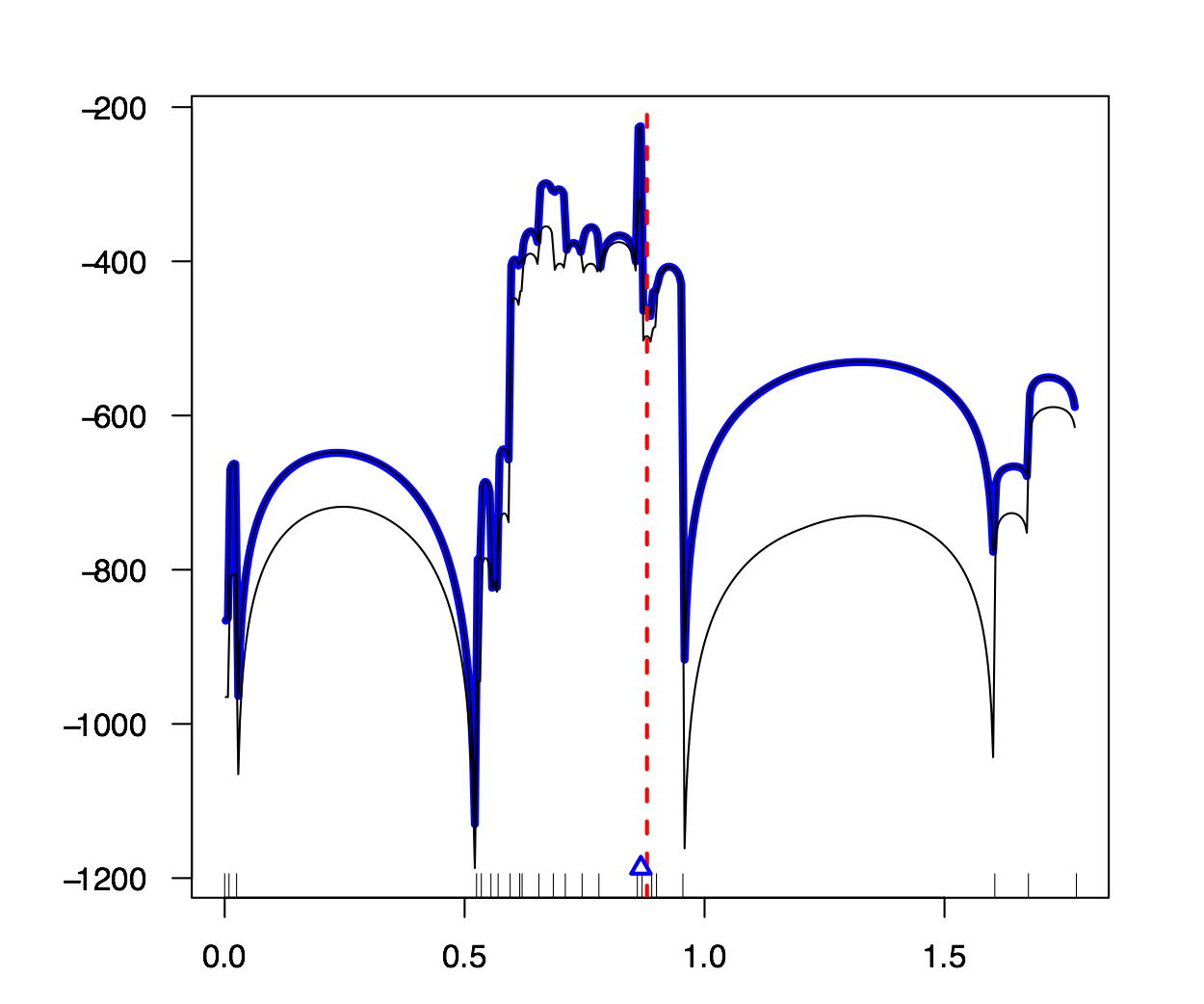
|
Natalia Dragieva (2008)
![]()
![]()
|
|
Construction d'un intervalle de confiance par la méthode bootstrap et test de permutation. Ce mémoire traite d'une application pratique de deux méthodes statistiques non paramétriques : le bootstrap et le test de permutation. La méthode du bootstrap a été proposée par Bradley Efron (1979) comme une alternative aux modèles mathématiques traditionnels dans des problèmes d'inférence complexe; celle-ci fournit plusieurs avantages sur les méthodes d'inférence traditionnelles. L'idée du test de permutation est apparue au début du XXème siècle dans les travaux de Neyman, Fisher et Pitman. Le test de permutation, très intensif quant au temps de calcul, est utilisé pour construire une distribution empirique de la statistique de test sous une hypothèse afin de la comparer avec la distribution de la même statistique sous l'hypothèse alternative.
Notre objectif est de déterminer l'intervalle de confiance pour un estimateur à maxi mum de vraisemblance d'une méthode de cartographie génétique existante (MapArg, Larribe et al. 2002) et de tester la qualité de cet estimateur, c'est-à-dire d'établir des seuils de significa tion pour la fonction de la vraisemblance. Les deux méthodes utilisent le calcul répétitif d'une même statistique de test sur des échantillons obtenus à partir de l'échantillon initial, soit avec le «bootstrap», soit avec des permutations. Dans un test d'hypothèse, les deux méthodes sont complémentaires.
Le but de ce mémoire est de proposer différentes variantes pour la construction de l'intervalle de confiance, et de tester des hypothèses distinctes, afin de trouver la meilleure solution adaptée pour la méthode MapArg. Pour faciliter la compréhension des décisions prises, un rappel de l'inférence statistique et des tests d' hypothèse est fait dans les chapitres 4 el 5 où la théorie du bootstrap et celle de test de permutation sont présentées. Comme les qualités d'un estimateur dépendent de la méthode utilisée pour le calculer, les chapitres 1 et 2 présentent la base biologique et la base en mathématiques sur lesquelles la méthode MapArg est construite, tandis qu'on trouvera dans le chapitre 3 une explication de la méthode MapArg.
|
|
|
|
Djamila Abed (2008)
![]()
|
|
Tests de déséquilibre de liaison et leur application à un gène candidat à l'hyperactivité. Ce mémoire présente deux tests génétiques permettant l'identification de gènes reliés à une maladie par l'intermédiaire des études d'association et de liaison génétique. Le premier test, le TDT (Transmission Test for Linkage disequilibrium), est très couramment utilisé et s'applique aux caractères binaires. Le second test, le QTDT est relativement nouveau et s'applique aux caractères mesurés sur une échelle continue. On commence par donner des notions théoriques de ces deux tests, en particulier des notions élémentaires de génétique quantitative. Par la suite, ces deux tests sont appliqués à des données réelles, des enfants atteints du TDA/H (trouble du déficit de l'attention, avec ou sans hyperactivité), et leurs résultats sont comparés. Les données comprennent des variables explicatives environnementales et génétiques. Les phénotypes (variables expliquées) sont des scores de comportement de l'enfant, qui varient sur une échelle continue. On montre que les résultats des deux tests sont similaires; le gène étudié n'est ni associé, ni lié au trouble étudié.
|
|
|
|
Karine Cyrenne (2006)
![]()
|
|
Rééchantillonnage de graphes de recombinaison ancestraux : aspects algorithmiques et statistiques. Ce mémoire traite d’une application statistique de la théorie de la coalescence en génétique des populations. Le processus de coalescence est un processus stochastique d’analyse en rétrospective de l’évolution de séquences de nucléotides d’une même espèce. Ce processus a été introduit par Kingman (1982) et nous utilisons un modèle mathématique qui est une variante enrichie de celui-ci car il incorpore les recombinaisons. Il s’agit du graphe de recombinaison ancestral (ARG) introduit et étudié par Griffiths et ses collaborateurs (1994, 1995 et 1997). À partir de ce modèle nous pouvons générer des graphes qui reconstituent l’évolution dans le temps de la population d’intérêt (nous retraçons la généalogie jusqu’à l’ancêtre commun, supposé unique).
La question pratique est d’identifier le site d’une mutation qui est responsable d’une maladie, à partir d’un échantillon de séquences provenant de sujets malades et sains. La méthodologie est largement une suite des travaux de Larribe et al. (2002). L’idée est d’insérer la maladie (virtuellement) à des sites spécifiques sur les séquences, de simuler un grand nombre de graphes ARG qui relient les séquences de l’échantillon, et de calculer la position la plus probable de la mutation d’intérêt. Ces simulations sont très coûteuses en temps d’exécution et en espace mémoire. De plus, il arrive qu’un assez grand nombre de graphes s’avère peu informatif.
Ainsi, le but de ce mémoire est de proposer une variante des algorithmes déjà connus, afin de mieux répondre aux problèmes mentionnés plus haut. Nous allons introduire préalablement plusieurs autres variantes que nous avons explorées avant de détailler celle que nous avons retenue. L’idée centrale a été suggérée par Liu (2000, 2005), soit d’éliminer tôt (bien avant d’arriver à l’ancêtre commun) les graphes qui apportent peu d’information. Nous avons proposé des solutions nouvelles en ce qui concerne le critère de choix des meilleurs graphes ainsi que pour l’implantation informatique. L’algorithme de base et ses variantes sont présentés aux chapitres 4 et 5, tandis que les autres chapitres introduisent d’une façon non technique les concepts probabilistes et biologiques qui sont nécessaires à la compréhension des algorithmes.
|
|
|
|
