
Cartographie génétique fine simultanée de deux gènes.
Mots-clés : statistique génétique, cartographie génétique, caractère polygénique, processus de coalescence, arbre de recombinaison ancestral.
Depuis maintenant une vingtaine d’années, la course aux gènes affectant certains caractères et maladies est ouverte ; de nouvelles technologies permettent le séquençage du génome humain; de grands projets à l’échelle planétaire accumulent les données génétiques. Plusieurs gènes ayant un impact sur certaines maladies ont été découverts à l’aide d’outils statistiques. Mais parfois, ces méthodes échouent face à certaines maladies complexes influencées simultanément par plusieurs gènes. C’est pourquoi le développement de méthodes de cartographie génétique adaptées à cette réalité est souhaitable.
L’objectif de ce mémoire est l’adaptation de la méthode de cartographie génétique fine MapArg (Larribe, Lessard et Schork, 2002 ; Larribe et Lessard, 2008) à la cartographie de caractère polygénique (c’est-à-dire influencé par plusieurs gènes). Nous supposerons que le caractère est causé uniquement par l’effet combiné de deux gènes et qu’il n’est pas influencé par des facteurs environnementaux. Bien que ces hypothèses posées sur la maladie paraîtront possiblement peu réalistes, elles nous permettront de simplifier la modélisation et de vérifier si nous sommes sur la bonne voie.
Pour permettre au lecteur non initié de comprendre les termes spécifiques à la génétique utilisés tout au long de ce mémoire, nous présentons au premier chapitre certains concepts de base de ce domaine. Nous poursuivons ensuite par une description de certains outils statistiques employés généralement en cartographie génétique, ce qui permettra au lecteur d’avoir un aperçu d’où, dans l’univers de la statistique génétique, s’inscrivent la méthode de cartographie MapArg et l’adaptation proposée de celle-ci. MapArg sera présentée en détail au chapitre trois. Suivra la présentation de l’adaptation de cette méthode à la cartographie de deux gènes causant un caractère. Nous terminerons ce mémoire, au chapitre cinq, avec la présentation des résultats obtenus lors de l’analyse de bases de données simulées.
Notre objectif était de proposer une adaptation de la méthode de cartographie génétique fine MapArg à la cartographie de caractère polygénique. Nous souhaitions aussi tester cette adaptation, établir ses limitations et la comparer aux méthodes de cartographie génétique existantes.
MapArg suppose que le caractère est influencé par un seul gène ; en utilisant un échantillon de cas et de contrôles, cette méthode estime la fonction de vraisemblance de la position de ce gène. Pour ce faire, elle doit inférer le génotype au gène cherché, à partir du phénotype, pour chacune des séquences de l’échantillon. Pour simplifier notre adaptation de MapArg à la cartographie de caractère polygénique, nous avons choisi de commencer par supposer que le caractère est influencé par deux gènes et d’utiliser un échantillon d’haplotypes ce qui, comparé aux diploïdes, facilite la modélisation. Nous devions modéliser l’interaction entre les gènes et déterminer comment ils affectent le caractère. De plus, il a fallu décider de la façon dont nous allions inférer les génotypes des TIM1 et TIM2 à partir des phénotypes.
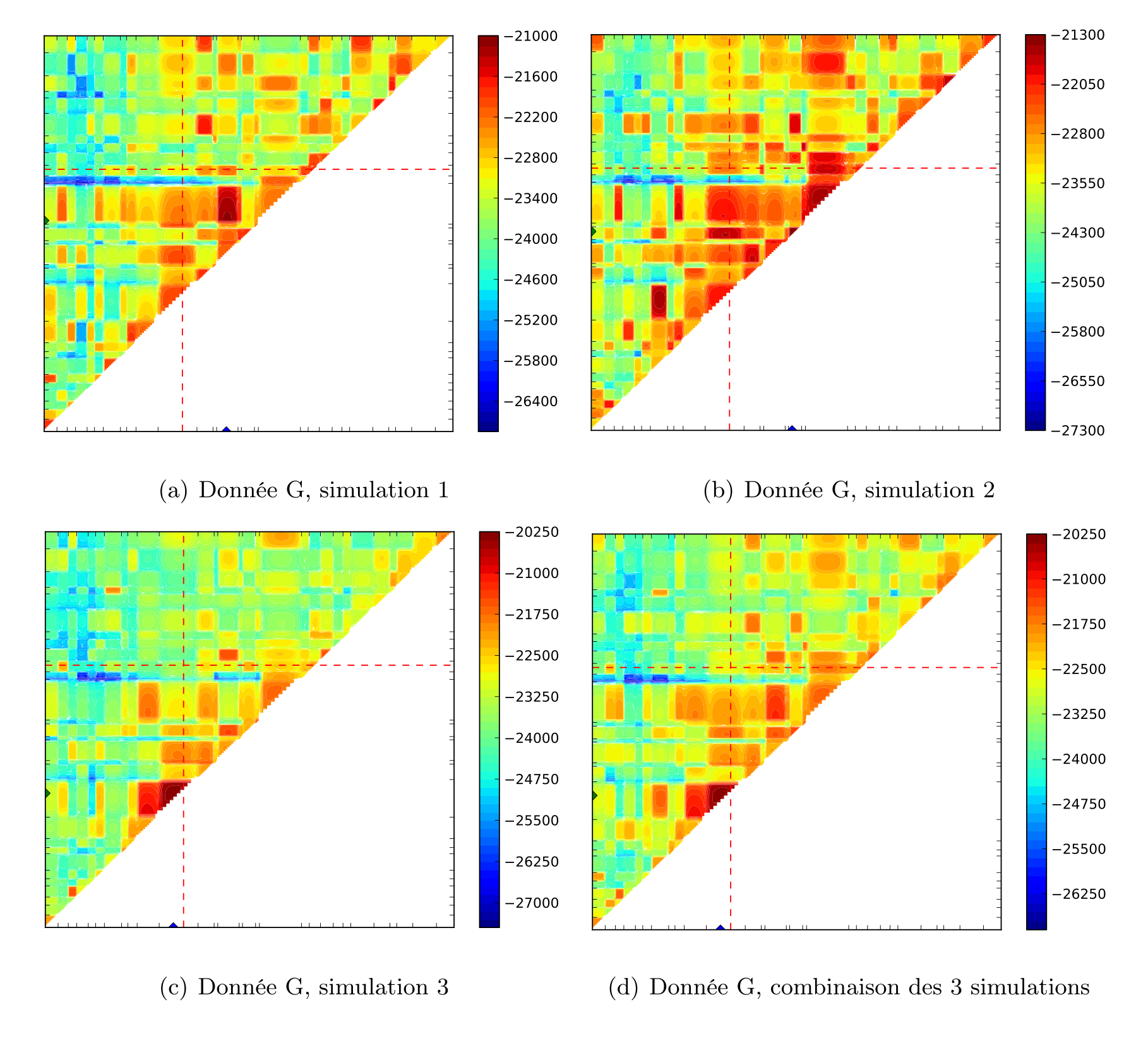
Des ensembles de données ont été simulés pour nous permettre de tester notre adaptation. Une séquence était affectée par le caractère si elle possédait deux allèles mutants, sinon cette séquence n’était pas affectée par le caractère. Pour l’inférence des génotypes à partir des phénotypes, nous avons choisi qu’une séquence affectée possèderait les deux allèles mutants, tandis qu’une séquence non atteinte du caractère possèderait deux allèles sains. Cela revient à supposer que la maladie est doublement récessive et les allèles mutants rares. Nous étions conscients de l’erreur d’inférence, mais nous ne pensions pas qu’elle aurait un impact sur l’estimation de la position des TIM1 et TIM2.
Les résultats obtenus avec la mesure d’association r2 et MapArg pour les ensembles de données simulées, ont montré la nécessité d’adapter les méthodes de cartographie à la réalité des caractères polygéniques, car souvent ces deux méthodes ne trouvaient pas un des deux marqueurs cherchés. Le programme PyArg nous a permis de tester notre adaptation avec des temps de calcul raisonnables. Les premiers résultats obtenus étaient tout de même encourageants, nous arrivions à trouver un des gènes causaux. Nous avons tenté de faire plus de graphes par intervalle, d’utiliser la vraisemblance composite pour tenter de diminuer la variabilité observée en faisant plusieurs répétitions de l’estimation de la vraisemblance.
C’est en utilisant les génotypes aux marqueurs cherchés pour l’inférence que nous avons obtenu les meilleurs résultats. L’erreur faite lors de l’inférence était donc significative. Bien sûr, d’autres facteurs pourraient améliorer notre estimation. L’utilisation d’une meilleure distribution proposée pour la construction des graphes pourrait aider. Mais pour éventuellement utiliser l’adaptation proposée, il faut trouver un moyen d’améliorer l’inférence des génotypes aux gènes causaux. Il faudrait aussi tester l’adaptation de MapArg avec différentes modélisations de l’interaction des gènes causaux.
