
ASSOCIATION GÉNÉTIQUE MESURÉE À L'AIDE D'UNE MÉTHODE DE CLASSIFICATION DE DONNÉES FONCTIONNELLES
Mots-clés : données fonctionnelles, semi-métriques, classification supervisée, SNP.
Grâce aux développements technologiques, on a maintenant accès à des instruments de mesure puissants qui permettent quasiment d’enregistrer des données en continu sur un intervalle de temps. On peut donc s’intéresser à des observations qui prennent par exemple, la forme de courbes, d’images ou de surfaces ; l’analyse de données fonctionnelles est une branche de la statistique permettant d’analyser de telles données.
L’objectif principal de ce mémoire est de proposer l’utilisation d’une méthode de classification supervisée comme outil d’association génétique. La méthode de classification que nous allons utiliser est une méthode non-paramétrique qui est basée sur l’estimateur de Nadaraya-Watson et developpée par Ferraty et Vieu (2006). Dans ce mémoire, nous allons expliquer comment le taux de classification à différents marqueurs génétiques peut être utilisé pour mesurer la force d’une as- sociation génétique, puis nous allons appliquer la méthode aux données génétiques d’une plante à fleurs de Moore et al. (2013) pour pouvoir décrire l’association entre le phénotype et le génotype. Ces études d’association ont pour but d’identifier les gènes sur plusieurs chromosomes influençant des caractères d’intérêt.
Le premier chapitre de ce mémoire contient une introduction des concepts de base en analyse de données fonctionnelles qui seront utilisés dans les chapitres subséquents. Le deuxième chapitre introduit les concepts de classification, de semi- métriques, l’estimateur des K plus proches voisins, l’estimation par noyau ainsi que la méthode de classification non-paramétrique pour des données fonction- nelles. Au chapitre trois, nous présentons une étude de simulation afin d’évaluer la performance de la méthode non-paramétrique de classification supervisé. Nous terminons ce mémoire, avec le quatrième chapitre, par la présentation de notre nouvelle méthode, puis nous présentons les résultats obtenus à l’aide de cette méthode, enfin nous les comparons à ceux de Kwak et al. (2016).
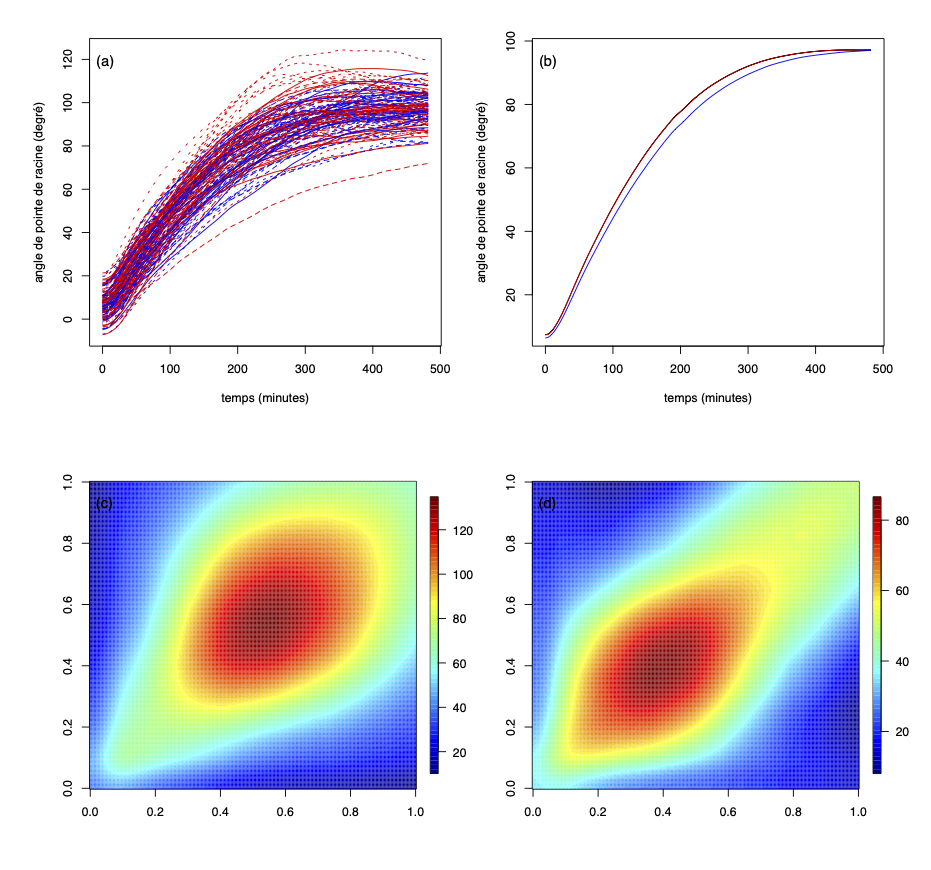
L’objectif de ce mémoire était de proposer une nouvelle approche d’association fonctionnelle utilisant des outils décrits dans Ferraty et Vieu (2006), et la comparer à l’approche par vraisemblance de Kwak et al. (2016) afin d’étudier l’association entre un caractère et des marqueurs génétiques. Nous avons évalué le comportement de la méthode par une étude de simulation. Nous avons choisi trois scénarios pour lesquels chaque scénario varie par rapport aux fonctions moyennes et fonctions de covariance. Pour chaque scénario, trois niveaux de difficulté de classification ont été proposés afin d’étudier les différentes combinaisons possibles de semi-métrique et du type de noyau.
Après avoir présenté notre nouvelle approche permettant de mesurer l’association entre génotype et phénotype, et l’analyse brute de nos données génétiques de Moore et al. (2013), nous avons utilisé les résultats obtenus avec l’étude de simulation du chapitre 3, pour sélectionner la semi-métrique basée sur les dérivées et le noyau triangle dans le but de trouver s’il y a une association entre le caractère d’intérêt et les marqueurs génétiques des données génétiques. Notre approche semble indiquer que les marqueurs génétiques les plus liés au caractère d’intérêt, sont sur les chromosomes 1 et 5. Après avoir comparé les résultats obtenus avec notre méthode proposée à ceux de Kwak et al. (2016), nous avons obtenu des résultats similaires à ceux de Kwak et al. (2016) pour les chromosomes 1 et 4, alors que pour les chromosomes 2,3 et 5, nous avons obtenu des résultats différents. Nous avons constaté que même sans avoir à effectuer des tests statistiques, nous avons eu des resultats très similaires à l’analyse quantitative multiple des locus de caractères quantitatifs pour les chromosomes 1 et 4. Ainsi, l’approche que nous proposons fonctionne assez pour poursuivre une étude plus approfondie dans cette voie.
